
Introduction
Sickle-cell anemia is an inherited autosomal linked recessive disease.
Sickle-cell anemia is a genetic blood disorder characterized by the production of abnormal hemoglobin, called hemoglobin S or sickle hemoglobin. Hemoglobin is a protein in red blood cells that carries oxygen throughout the body. In sickle cell anemia, the abnormal hemoglobin causes red blood cells to become rigid, sticky, and shaped like sickles or crescent moons rather than the usual round shape.
Symptoms of sickle cell anemia
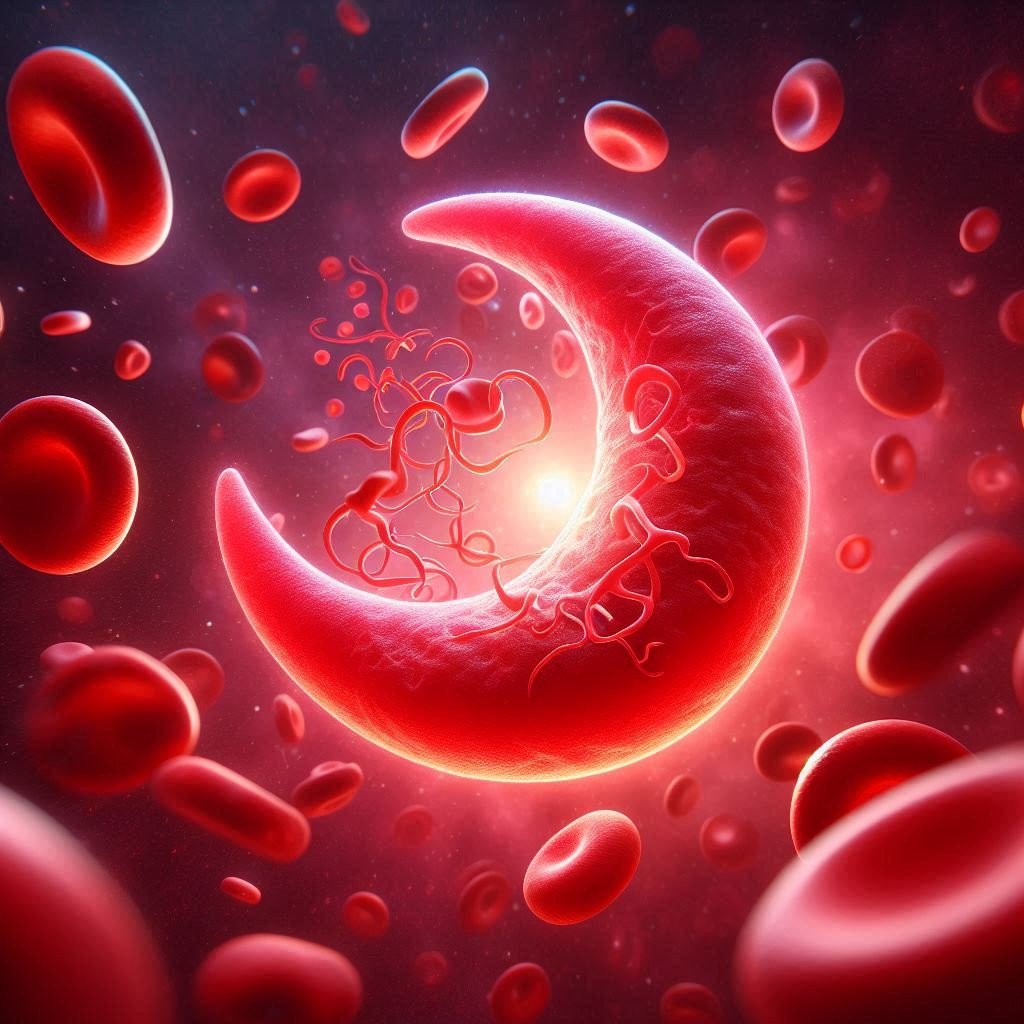
1. Pain Events (Sickle Cell Crisis): Pain episodes, sometimes referred to as sickle cell crises, are one of the main signs and symptoms of sickle cell anemia. Sickle-shaped red blood cells obstruct blood flow to tissues and organs, causing these events. Although pain can occur anywhere in the body, it frequently affects the joints, bones, chest, and abdomen.
2. Anemia: Sickle cell anemia often leads to chronic anemia due to the reduced lifespan of sickle-shaped red blood cells. Anemia can cause fatigue, weakness, and shortness of breath.
3. Fatigue: Chronic anemia and the body’s increased demand for oxygen can result in fatigue and decreased energy levels.
4. Jaundice: Jaundice, characterized by yellowing of the skin and eyes, can occur due to the breakdown of red blood cells and the release of bilirubin into the bloodstream.
5. Swelling of Hands and Feet (Dactylitis): In infants and young children with sickle cell anemia, dactylitis, or swelling of the hands and feet, may occur.
6. Frequent Infections: Sickle cell anemia can impair the function of the spleen, increasing the risk of infections, particularly with encapsulated bacteria such as Streptococcus pneumoniae.
7. Delayed Growth or Development: Children with sickle cell anemia may experience delayed growth and development.
8. Vision Problems: Sickle cell anemia can damage the blood vessels in the eyes, leading to vision problems or even vision loss.
9. Organ Damage: Prolonged or recurrent episodes of reduced blood flow can lead to organ damage over time. Organs commonly affected include the spleen, liver, lungs, kidneys, and brain.
10. Stroke: Sickle cell anemia increases the risk of stroke, particularly in children, due to blockages in the blood vessels supplying the brain.
11. Acute Chest Syndrome: Acute chest syndrome is a potentially life-threatening complication characterized by chest pain, fever, cough, and difficulty breathing. It can result from blockages in the blood vessels supplying the lungs.
12. Priapism: In males, sickle cell anemia can cause priapism, a painful and prolonged erection unrelated to sexual stimulation.
Causes

A genetic mutation in the gene encoding the hemoglobin beta-globin component, a protein present in red blood cells, results in sickle cell anemia. Red blood cells exhibit the distinctive sickling caused by hemoglobin S (HbS), an aberrant hemoglobin produced as a result of this mutation. Getting two copies of the defective gene—one from each parent—is the main cause of sickle cell anemia. We refer to this pattern of inheritance as autosomal recessive.
The disease is controlled by a single pair of allele, HbA and HbS Out of the three possible genotypes only homozygous individuals for HbS (HbS HbS) show the diseased phenotype.
The disorder is caused by the substitution of Glutamic acid (Glu) by Valine (Val) at the 6th position of the beta globin chain of the hemoglobin molecule. Therefore it is an example of point substitution as on 6th codon of beta globin chain is from GAG to GUG.
Treatment
1. Pain Management:
- During emergencies, pain can be managed with the use of opioids, nonsteroidal anti-inflammatory medications (NSAIDs), and other analgesics.
- Hydration: Getting enough fluids is important to avoid dehydration, which can lead to sickle cell crises.
- Heat therapy: Relief from painful places can be achieved by applying heat.
2. Hydroxyurea Therapy:
- One drug that helps lessen sickle cell anemia-related problems and pain episodes’ frequency and intensity is hydroxyurea. It functions by raising the levels of fetal hemoglobin (HbF), which prevents sickle hemoglobin from polymerizing.
- Hydroxyurea may also help lower the risk of acute chest syndrome and other problems, as well as the requirement for blood transfusions.
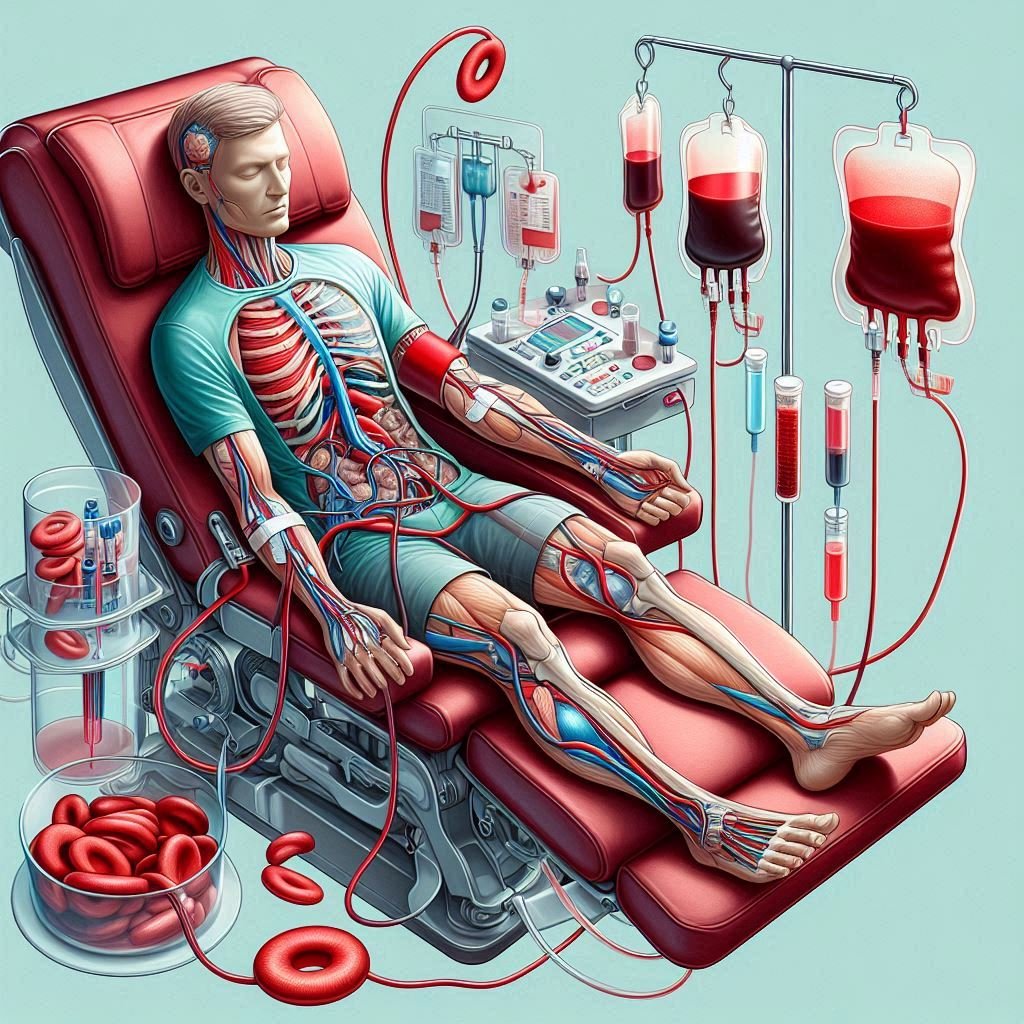
3. Blood Transfusions:
- Blood transfusions may be necessary to treat severe anemia or to prevent and manage complications such as stroke, acute chest syndrome, or severe organ damage.
- Chronic transfusions may also be used in certain cases to reduce the risk of recurrent strokes in individuals with a history of stroke.
4. Bone Marrow or Stem Cell Transplant:
- The only treatment currently available for sickle cell anemia is bone marrow or stem cell transplantation. The patient’s bone marrow, which generates faulty red blood cells, is being replaced during this treatment with healthy bone marrow stem cells from a suitable donor.
- Usually, people with severe sickle cell disease who have a suitable donor and have not responded to various forms of treatment are the ones who get stem cell transplants.
5. Management of Complications:
- Regular medical monitoring and management are essential to prevent and treat complications associated with sickle cell anemia, such as infections, acute chest syndrome, stroke, pulmonary hypertension, and organ damage.
- Vaccinations, antibiotic prophylaxis, and prompt treatment of infections are important preventive measures.
Precautions
Stay Hydrated: Drink plenty of fluids, especially water, to prevent dehydration, which can trigger sickle cell crises.
Avoid Extreme Temperatures: Extreme temperatures, both hot and cold, can trigger sickle cell crises. Stay indoors during extremely hot or cold weather, and dress appropriately for the weather.

Prevent Infections:
- Practice good hygiene, including regular handwashing, to reduce the risk of infections.
- Stay up to date with vaccinations, including the annual flu vaccine and vaccines for pneumonia and other preventable infections.
- Avoid close contact with individuals who have contagious illnesses, such as colds or the flu.
Manage Stress: Stress can trigger sickle cell crises in some individuals. Practice stress-reduction techniques such as deep breathing, meditation, yoga, or talking to a therapist or counselor.
Avoid High Altitudes: High altitudes can reduce oxygen levels in the blood, which can trigger sickle cell crises. Avoid travel to high-altitude areas if possible, and take precautions if travel is necessary.
Regular Medical Check-ups: Attend regular check-ups with healthcare providers experienced in the management of sickle cell disease. These check-ups are essential for monitoring the condition, managing symptoms, and preventing complications.

Take Medications as Prescribed: Take medications prescribed by healthcare providers as directed, including pain medications, hydroxyurea, and any other prescribed treatments.
Monitor Symptoms: Be vigilant for signs and symptoms of sickle cell crises, such as pain, fever, swelling, or difficulty breathing. Seek medical attention promptly if symptoms worsen or new symptoms develop.
Maintain a Healthy Lifestyle: Eat a balanced diet, get regular exercise (within limits recommended by healthcare providers), and get enough sleep to support overall health and well-being.
Genetic Counseling: Consider genetic counseling if planning to have children. Genetic counseling can help individuals understand their risk of passing sickle cell disease to their children and explore options for family planning.

FAQ (frequently asked questions)
Q. What causes sickle cell anemia?
Ans. Sickle cell anemia is caused by inheriting two copies of the mutated hemoglobin S gene, one from each parent. It is an autosomal recessive genetic disorder.
Q. What are the symptoms of sickle cell anemia?
Ans. Common symptoms include pain episodes (sickle cell crises), anemia, fatigue, jaundice, swelling of hands and feet (dactylitis), frequent infections, delayed growth and development, vision problems, organ damage, stroke, acute chest syndrome, and priapism (prolonged and painful erection in males).
Q. How is sickle cell anemia diagnosed?
Ans. Sickle cell anemia is diagnosed through blood tests, including hemoglobin electrophoresis, which identifies abnormal hemoglobin variants. Newborn screening programs are also in place in many countries to detect sickle cell disease early.
Q. What are the treatment options for sickle cell anemia?
Ans. Treatment aims to manage symptoms, prevent complications, and improve quality of life. Options include pain management, hydroxyurea therapy, blood transfusions, bone marrow or stem cell transplantation (in select cases), and supportive care measures.
Q. Can sickle cell anemia be cured?
Ans. Currently, there is no cure for sickle cell anemia. However, bone marrow or stem cell transplantation offers the potential for a cure in select cases.
Q. What lifestyle changes can help manage sickle cell anemia?
Ans. Staying hydrated, avoiding extreme temperatures, preventing infections, managing stress, maintaining a healthy lifestyle (including diet and exercise), and seeking regular medical care are important for managing sickle cell anemia.
Q. What is the life expectancy for individuals with sickle cell anemia?
Ans. Life expectancy for individuals with sickle cell anemia has improved significantly in recent decades with advances in treatment and supportive care. With proper management, many individuals can live well into adulthood.
Q. Is sickle cell anemia contagious?
Ans. No, sickle cell anemia is not contagious. It is an inherited genetic disorder caused by mutations in specific genes.
Conclusion
Sickle cell anemia is a complex genetic disorder that requires comprehensive management to optimize outcomes and quality of life for affected individuals. While there is currently no cure, advances in research and medical technology offer hope for improved treatments and potential cures in the future. By raising awareness, promoting early diagnosis, and providing access to multidisciplinary care, we can empower individuals with sickle cell anemia to live fulfilling lives despite the challenges posed by this condition.
Table of Contents
Read more about sickle cell anemia
Go and visit dusearchit.in and get more knowledge about others topics.
Pingback: How dangerous is phenylketonuria ? symptoms.(2024)